Validation¶
Validation methodology¶
In this section, a force field is constructed using the current version
of QuickFF (i.e. 2.2.2), after which it is tested and evaluated for its
ability to reproduce the ab initio input it was fitted to. The force field was
constructed using the QuickFF script qff.py. The
required ab initio input is contained in the file gaussian.fchk and the
electrostatic partitioning in gaussian_mbis.h5. The Yaff parameter file for
the electrostatic contribution is generated using
qff-input-ei.py -f –ffatypes=low –gaussian gaussian.fchk gaussian_mbis.h5:charges pars_ei_mbisgauss.txt
The config file containing the settings that are used for the force field is
given by config.txt and can be viewed below. Furthermore, all required input
files as well as the generated output can be found in the directory
share/validation.
#IO
fn_yaff : pars_cov.txt
fn_charmm22_prm : None
fn_charmm22_psf : None
fn_sys : system.chk
plot_traj : final
xyz_traj : False
fn_traj : None
log_level : medium
log_file : None
#Program
program_mode : DeriveFF
#FF config
only_traj : PT_ALL
ffatypes : low
ei : pars_ei_mbisgauss.txt
ei_rcut : None #default is 20 (periodic) or 50 (non-per) A
vdw : None
vdw_rcut : 20*angstrom
covres : None
excl_bonds : None
excl_bends : None
excl_dihs : None
excl_oopds : None
do_hess_mass_weighting : True
do_hess_negfreq_proj : False
do_cross_svd : True
pert_traj_tol : 1e-9
cross_svd_rcond : 1e-9
do_bonds : True
do_bends : True
do_dihedrals : True
do_oops : True
do_cross_ASS : True
do_cross_ASA : True
do_cross_DSS : False
do_cross_DSD : False
do_cross_DAA : False
do_cross_DAD : False
bond_term : BondHarm
bend_term : BendAHarm
do_squarebend : True
do_bendclin : True
do_sqoopdist_to_oopdist : True
To evaluate the performance of the force field, it is applied to perform a basic geometry optimization followed by a normal mode analysis using Yaff and TAMkin. Finally, two validations are performed for each system:
the force field energy is compared to the ab initio energy along the perturbation trajectories that were constructed by QuickFF.
the geometry (i.e. bond lengths, bending angles, dihedral angles and ut-of-plane distances) and normal mode frequencies as predicted by the force field are compared with the given ab initio input. To this end, the following statistics are used:
Root Mean Square Deviation (RMSD)
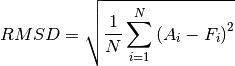
Mean Deviation (RMSD)
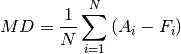
Root Mean Square Error (RMSE)
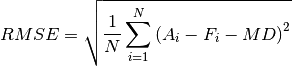
These statistics satisfy the condition
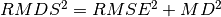 .
.
Validation 1 - water¶
Force field construction¶
Averaged Atomic Charges:
------------------------
H 0.451 +- 0.000 (N=2)
O -0.901 +- 0.000 (N=1)
#Fixed charges
#---------------
FIXQ:UNIT Q0 e
FIXQ:UNIT P e
FIXQ:UNIT R angstrom
FIXQ:SCALE 1 1.0
FIXQ:SCALE 2 1.0
FIXQ:SCALE 3 1.0
FIXQ:DIELECTRIC 1.0
# Atomic parameters
# ----------------------------------------------------
# KEY label Q_0A R_A
# ----------------------------------------------------
FIXQ:ATOM H 0.4506107006 0.7308000000
FIXQ:ATOM O -0.9012712115 1.1325000000
________________/\\\_________/\\\\\\\\\\\\\\\__/\\\\\\\\\\\\\\\_________________
______________/\\\\/\\\\_____\/\\\///////////__\/\\\///////////_________________
_____________/\\\//\////\\\___\/\\\_____________\/\\\___________________________
_____________/\\\______\//\\\__\/\\\\\\\\\\\_____\/\\\\\\\\\\\__________________
_____________\//\\\______/\\\___\/\\\///////______\/\\\///////__________________
_______________\///\\\\/\\\\/____\/\\\_____________\/\\\________________________
__________________\////\\\//______\/\\\_____________\/\\\_______________________
______________________\///\\\\\\___\/\\\_____________\/\\\______________________
_________________________\//////____\///______________\///______________________
Welcom to QuickFF
a Python package to quickly derive force fields from ab initio input data
Written by
Louis Vanduyfhuys(1)*, Steven Vandenbrande(1) and Toon Verstraelen(1)
(1) Center for Molecular Modeling, Ghent University Belgium.
* mailto: Louis.Vanduyfhuys@UGent.be
USER louis
MACHINE Linux T085 4.15.0-39-generic #42-Ubuntu SMP Tue Oct 23
MACHINE 15:48:01 UTC 2018 x86_64
TIME 2018-11-29 16:23:19.571237
QUICKFF VERSION 2.2.1
PYTHON VERSION 3.6.3 |Anaconda custom (64-bit)| (default, Oct 13 2017,
PYTHON VERSION 12:02:49) [GCC 7.2.0]
NUMPY VERSION 1.13.3
SCIPY VERSION 0.19.1
MATPLOTLIB VERSION 2.1.0
CURRENT DIR /home/louis/build/quickff/share/validation/water
COMMAND LINE /home/louis/build/anaconda3/bin/qff.py -c ../config.txt
COMMAND LINE gaussian.fchk
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
INIT Initializing system
INIT Initializing Second order taylor reference for ai
INIT Initializing Yaff force field reference for EI
INIT Initializing program
VAL Initializing valence force field
VAL Added 2 bond terms
VAL Added 1 bend terms (an)harmonic in the angle, 0 bend terms with
VAL 1+cos(angle) potential and 0 bend terms with 1-cos(4*theta) potential.
VAL Added 0 Harmonic and 0 SquareHarmonic out-of-plane distance terms
EQSET Resetting system coordinates to ab initio ref
EQSET Setting rest values to AI equilibrium values for tasks EQ_RV
EQSET Averaging force field parameters over master and slaves
PTGEN Resetting system coordinates to ab initio ref
PTGEN Constructing trajectories
PTEST Resetting system coordinates to ab initio ref
PTEST Estimating FF parameters from perturbation trajectories
PTPOST Averaging force field parameters over master and slaves
VAL Resetting system coordinates to ab initio ref
HCEST Resetting system coordinates to ab initio ref
HCEST Estimating force constants from Hessian cost for tasks HC_FC_DIAG HC_FC_
HCEST CROSS_ASS HC_FC_CROSS_ASA
PTEST Resetting system coordinates to ab initio ref
PTEST Estimating FF parameters from perturbation trajectories with valence
PTEST reference
PTPOST Averaging force field parameters over master and slaves
HCEST Resetting system coordinates to ab initio ref
HCEST Estimating force constants from Hessian cost for tasks HC_FC_DIAG HC_FC_
HCEST CROSS_ASS HC_FC_CROSS_ASA
HCEST Resetting system coordinates to ab initio ref
HCEST Estimating force constants from Hessian cost for tasks HC_FC_CROSS_ASS
HCEST HC_FC_CROSS_ASA HC_FC_CROSS_DSS HC_FC_CROSS_DSD HC_FC_CROSS_DAA HC_FC_
HCEST CROSS_DAD
BondHarm/H.O (kjmol/A^2 A)
fc = 4979 +- .000 rv = .9767 +- .000
BendAHarm/H.O.H (kjmol/rad^2 deg)
fc = 346.5 +- .000 rv = 95.53 +- .000
Cross/H.O.H/b0a (kjmol/A A deg)
fc = 145 +- .0 rv0 = .971 +- 0.0 rv1 = 96.0 +- 0.0
Cross/H.O.H/b1a (kjmol/A A deg)
fc = 145 +- .0 rv0 = .971 +- 0.0 rv1 = 96.0 +- 0.0
Cross/H.O.H/bb (kjmol/A^2 A A)
fc = -110 +- .0 rv0 = .971 +- 0.0 rv1 = .971 +- 0.0
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
TIMING Initializing 0:00:00.041719
TIMING Equil Set RV 0:00:00.000183
TIMING PT Generate 0:00:00.210871
TIMING PT Estimate 0:00:00.012061
TIMING PT Post process 0:00:00.000409
TIMING HC Estimate FC 0:00:00.031958
TIMING PT plot energy 0:00:00.514098
__/\\\__________________________________________________________________/\\\____
\ \\\ \ \\\
\ \\\ End of file. Thanks for using QuickFF! Come back soon!! \ \\\
____\///__________________________________________________________________\///__
# BONDHARM
#---------
BONDHARM:UNIT K kjmol/A**2
BONDHARM:UNIT R0 A
BONDHARM:PARS H O 4.9793007398e+03 9.7671602900e-01
# BENDAHARM
#----------
BENDAHARM:UNIT K kjmol/rad**2
BENDAHARM:UNIT THETA0 deg
BENDAHARM:PARS H O H 3.4652118756e+02 9.5532795842e+01
# Cross
#------
Cross:UNIT KSS kjmol/angstrom**2
Cross:UNIT KBS0 kjmol/(angstrom*rad)
Cross:UNIT KBS1 kjmol/(angstrom*rad)
Cross:UNIT R0 angstrom
Cross:UNIT R1 angstrom
Cross:UNIT THETA0 deg
Cross:PARS H O H -1.1006640167e+02 1.4564327376e+02 1.4564327376e+02 9.7199818096e-01 9.7199818096e-01 9.6002094143e+01
Force field evaluation¶
Perturbation trajectories¶
The figures below illustrate the reproduction of the ab initio energy along the constructed perturbation trajectories. For water, there are 3 such trajectories, two bonds and one bend.
- First O-H bond:

- Second O-H bond:

- H-O-H bend:

Geometry and frequencies¶
OBSERVABLE [UNIT] | RMSD | MD | RMSE
--------------------|----------------|----------------|----------------
| | |
GEOMETRY | | |
Bonds [A] | 7.214e-05 | -7.214e-05 | 0.000e+00
Bends [deg] | 5.485e-02 | 5.485e-02 | 0.000e+00
Dihedrals [deg] | - | - | -
Opdists [A] | - | - | -
| | |
FREQUENCIES [1/cm] | | |
All | 7.656e-02 | -2.408e-02 | 7.267e-02
0 - 100 | - | - | -
100 - 500 | - | - | -
500 - 1000 | - | - | -
1000 - 3000 ( 1) | 7.841e-02 | 7.841e-02 | 0.000e+00
3000 - inf ( 2) | 7.561e-02 | -7.533e-02 | 6.554e-03
Validation 2 - benzene¶
Force field construction¶
Averaged Atomic Charges:
------------------------
H 0.131 +- 0.000 (N=6)
C -0.132 +- 0.001 (N=6)
#Fixed charges
#---------------
FIXQ:UNIT Q0 e
FIXQ:UNIT P e
FIXQ:UNIT R angstrom
FIXQ:SCALE 1 1.0
FIXQ:SCALE 2 1.0
FIXQ:SCALE 3 1.0
FIXQ:DIELECTRIC 1.0
# Atomic parameters
# ----------------------------------------------------
# KEY label Q_0A R_A
# ----------------------------------------------------
FIXQ:ATOM H 0.1314377213 0.7308000000
FIXQ:ATOM C -0.1315514661 1.1703000000
________________/\\\_________/\\\\\\\\\\\\\\\__/\\\\\\\\\\\\\\\_________________
______________/\\\\/\\\\_____\/\\\///////////__\/\\\///////////_________________
_____________/\\\//\////\\\___\/\\\_____________\/\\\___________________________
_____________/\\\______\//\\\__\/\\\\\\\\\\\_____\/\\\\\\\\\\\__________________
_____________\//\\\______/\\\___\/\\\///////______\/\\\///////__________________
_______________\///\\\\/\\\\/____\/\\\_____________\/\\\________________________
__________________\////\\\//______\/\\\_____________\/\\\_______________________
______________________\///\\\\\\___\/\\\_____________\/\\\______________________
_________________________\//////____\///______________\///______________________
Welcom to QuickFF
a Python package to quickly derive force fields from ab initio input data
Written by
Louis Vanduyfhuys(1)*, Steven Vandenbrande(1) and Toon Verstraelen(1)
(1) Center for Molecular Modeling, Ghent University Belgium.
* mailto: Louis.Vanduyfhuys@UGent.be
USER louis
MACHINE Linux T085 4.15.0-39-generic #42-Ubuntu SMP Tue Oct 23
MACHINE 15:48:01 UTC 2018 x86_64
TIME 2018-11-29 16:23:22.266821
QUICKFF VERSION 2.2.1
PYTHON VERSION 3.6.3 |Anaconda custom (64-bit)| (default, Oct 13 2017,
PYTHON VERSION 12:02:49) [GCC 7.2.0]
NUMPY VERSION 1.13.3
SCIPY VERSION 0.19.1
MATPLOTLIB VERSION 2.1.0
CURRENT DIR /home/louis/build/quickff/share/validation/benzene
COMMAND LINE /home/louis/build/anaconda3/bin/qff.py -c ../config.txt
COMMAND LINE gaussian.fchk
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
INIT Initializing system
INIT Initializing Second order taylor reference for ai
INIT Initializing Yaff force field reference for EI
INIT Initializing program
VAL Initializing valence force field
VAL Added 12 bond terms
VAL Added 18 bend terms (an)harmonic in the angle, 0 bend terms with
VAL 1+cos(angle) potential and 0 bend terms with 1-cos(4*theta) potential.
VAL Added 6 Harmonic and 0 SquareHarmonic out-of-plane distance terms
EQSET Resetting system coordinates to ab initio ref
EQSET Setting rest values to AI equilibrium values for tasks EQ_RV
EQSET Averaging force field parameters over master and slaves
PTGEN Resetting system coordinates to ab initio ref
PTGEN Constructing trajectories
PTEST Resetting system coordinates to ab initio ref
PTEST Estimating FF parameters from perturbation trajectories
PTPOST Averaging force field parameters over master and slaves
VAL Resetting system coordinates to ab initio ref
HCEST Resetting system coordinates to ab initio ref
HCEST Estimating force constants from Hessian cost for tasks HC_FC_DIAG HC_FC_
HCEST CROSS_ASS HC_FC_CROSS_ASA
PTEST Resetting system coordinates to ab initio ref
PTEST Estimating FF parameters from perturbation trajectories with valence
PTEST reference
PTPOST Averaging force field parameters over master and slaves
HCEST Resetting system coordinates to ab initio ref
HCEST Estimating force constants from Hessian cost for tasks HC_FC_DIAG HC_FC_
HCEST CROSS_ASS HC_FC_CROSS_ASA
HCEST Resetting system coordinates to ab initio ref
HCEST Estimating force constants from Hessian cost for tasks HC_FC_CROSS_ASS
HCEST HC_FC_CROSS_ASA HC_FC_CROSS_DSS HC_FC_CROSS_DSD HC_FC_CROSS_DAA HC_FC_
HCEST CROSS_DAD
BondHarm/C.C (kjmol/A^2 A)
fc = 3965 +- .000 rv = 1.394 +- .000
BondHarm/C.H (kjmol/A^2 A)
fc = 3333 +- .000 rv = 1.087 +- .000
BendAHarm/C.C.C (kjmol/rad^2 deg)
fc = 601.9 +- .000 rv = 119.9 +- .000
BendAHarm/C.C.H (kjmol/rad^2 deg)
fc = 320.9 +- .000 rv = 120.1 +- .000
TorsCheby2/C.C.C.C (kjmol au)
fc = 35.9 +- 0.0 sgn = -1
TorsCheby2/C.C.C.H (kjmol au)
fc = 31.4 +- 0.0 sgn = -1
TorsCheby2/H.C.C.H (kjmol au)
fc = 16.8 +- .00 sgn = -1
Oopdist/C.C.H.C (kjmol/A^2 A)
fc = 166.2 +- .000 rv = .0000 +- .000
Cross/C.C.C/b0a (kjmol/A A deg)
fc = 49.2 +- .0 rv0 = 1.39 +- 0.0 rv1 = 120 +- 0.0
Cross/C.C.C/b1a (kjmol/A A deg)
fc = 49.3 +- .0 rv0 = 1.39 +- 0.0 rv1 = 120 +- 0.0
Cross/C.C.C/bb (kjmol/A^2 A A)
fc = 525 +- .0 rv0 = 1.39 +- 0.0 rv1 = 1.39 +- 0.0
Cross/C.C.H/b0a (kjmol/A A deg)
fc = 113 +- .0 rv0 = 1.39 +- .00 rv1 = 120 +- .00
Cross/C.C.H/b1a (kjmol/A A deg)
fc = 139 +- .0 rv0 = 1.08 +- .00 rv1 = 120 +- .00
Cross/C.C.H/bb (kjmol/A^2 A A)
fc = 40.3 +- .0 rv0 = 1.39 +- .00 rv1 = 1.08 +- .00
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
TIMING Initializing 0:00:00.109844
TIMING Equil Set RV 0:00:00.001019
TIMING PT Generate 0:00:06.375875
TIMING PT Estimate 0:00:00.159069
TIMING PT Post process 0:00:00.003885
TIMING HC Estimate FC 0:00:00.346235
TIMING PT plot energy 0:00:05.643349
__/\\\__________________________________________________________________/\\\____
\ \\\ \ \\\
\ \\\ End of file. Thanks for using QuickFF! Come back soon!! \ \\\
____\///__________________________________________________________________\///__
# BONDHARM
#---------
BONDHARM:UNIT K kjmol/A**2
BONDHARM:UNIT R0 A
BONDHARM:PARS C H 3.3330947115e+03 1.0876030745e+00
BONDHARM:PARS C C 3.9651511136e+03 1.3940024805e+00
# BENDAHARM
#----------
BENDAHARM:UNIT K kjmol/rad**2
BENDAHARM:UNIT THETA0 deg
BENDAHARM:PARS C C H 3.2097790994e+02 1.2015408782e+02
BENDAHARM:PARS C C C 6.0195059267e+02 1.1997128609e+02
# TORSION
#--------
TORSION:UNIT A kjmol
TORSION:UNIT PHI0 deg
TORSION:PARS C C C H 2 3.1428590550e+01 0.0000000000e+00
TORSION:PARS C C C C 2 3.5982599978e+01 0.0000000000e+00
TORSION:PARS H C C H 2 1.6841710927e+01 0.0000000000e+00
# OOPDIST
#--------
OOPDIST:UNIT K kjmol/A**2
OOPDIST:UNIT D0 A
OOPDIST:PARS C C H C 1.6627861970e+02 4.7922360401e-17
# Cross
#------
Cross:UNIT KSS kjmol/angstrom**2
Cross:UNIT KBS0 kjmol/(angstrom*rad)
Cross:UNIT KBS1 kjmol/(angstrom*rad)
Cross:UNIT R0 angstrom
Cross:UNIT R1 angstrom
Cross:UNIT THETA0 deg
Cross:PARS C C H 4.0314250860e+01 1.1314941661e+02 1.3908938734e+02 1.3934733019e+00 1.0875970644e+00 1.2001586285e+02
Cross:PARS C C C 5.2510973026e+02 4.9272567406e+01 4.9272567406e+01 1.3934733019e+00 1.3934733019e+00 1.2003594733e+02
Force field evaluation¶
Perturbation trajectories¶
The figures below illustrate the reproduction of the ab initio energy along the constructed perturbation trajectories. For water, there are 3 such trajectories, two bonds and one bend.
- C-H bonds:






- C-C bonds:






- C-C-C bends:






- C-C-H bends:






- C-C-H-C out-of-plane distances:






Geometry and frequencies¶
OBSERVABLE [UNIT] | RMSD | MD | RMSE
--------------------|----------------|----------------|----------------
| | |
GEOMETRY | | |
Bonds [A] | 3.530e-04 | -2.392e-04 | 2.596e-04
Bends [deg] | 9.492e-08 | 7.105e-15 | 9.492e-08
Dihedrals [deg] | 5.371e-16 | 1.643e-32 | 5.371e-16
Opdists [A] | 5.587e-18 | 4.747e-18 | 2.947e-18
| | |
FREQUENCIES [1/cm] | | |
All | 2.338e+01 | 1.057e+00 | 2.335e+01
0 - 100 | - | - | -
100 - 500 ( 2) | 1.701e+01 | 1.700e+01 | 4.160e-01
500 - 1000 ( 8) | 1.470e+01 | -6.089e+00 | 1.338e+01
1000 - 3000 ( 14) | 3.163e+01 | 3.301e+00 | 3.146e+01
3000 - inf ( 6) | 3.739e+00 | 3.573e-02 | 3.739e+00