QuickFF literature¶
QuickFF: A Program for a Quick and Easy Derivation of Force Fields for Metal-Organic Frameworks from Ab Initio Input
Authors: L. Vanduyfhuys, S. Vandenbrande, T. Verstraelen, R. Schmid, M. Waroquier, V. Van Speybroeck
Journal: Journal of Computational Chemistry, 2015
Volume 36, Issue 13, Pages 1015-1027
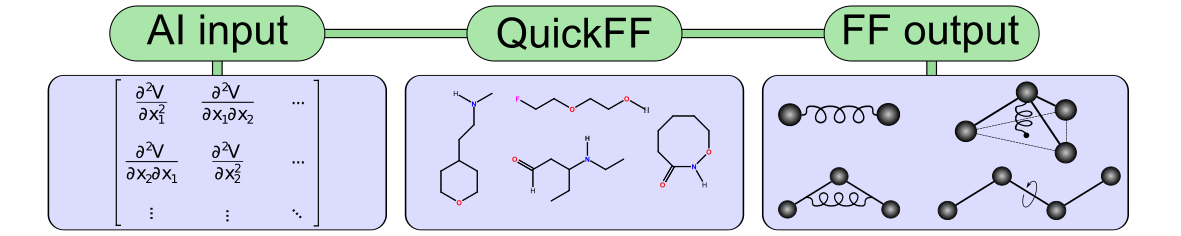
QuickFF is a software package to derive accurate force fields for isolated and complex molecular systems in a quick and easy manner. Apart from its general applicability, the program has been designed to generate force fields for metal-organic frameworks in an automated fashion. The force field parameters for the covalent interaction are derived from ab initio data. The mathematical expression of the covalent energy is kept simple to ensure robustness and to avoid fitting deficiencies as much as possible. The user needs to produce an equilibrium structure and a Hessian matrix for one or more building units. Afterwards, a force field is generated for the system using a three-step method implemented in QuickFF. The first two steps of the methodology are designed to minimize correlations among the force field parameters. In the last step the parameters are refined by imposing the force field parameters to reproduce the ab initio Hessian matrix in Cartesian coordinate space as accurate as possible. The method is applied on a set of 1000 organic molecules to show the easiness of the software protocol. To illustrate its application to MOFs, QuickFF is used to determine force fields for MIL-53(Al) and MOF-5. For both materials accurate force fields were already generated in literature but they requested a lot of manual interventions. QuickFF is a tool that can easily be used by anyone with a basic knowledge of performing ab initio calculations. As a result accurate force fields are generated with minimal effort.
Extension of the QuickFF Force Field Protocol for an Improved Accuracy of Structural, Vibrational, Mechanical and Thermal Properties of Metal-Organic Frameworks
Authors: L. Vanduyfhuys, S. Vandenbrande, J. Wieme, M. Waroquier, T. Verstraelen, V. Van Speybroeck
Journal: Journal of Computational Chemistry, 2018
Volume 39, Issue 16, Pages 999-1011
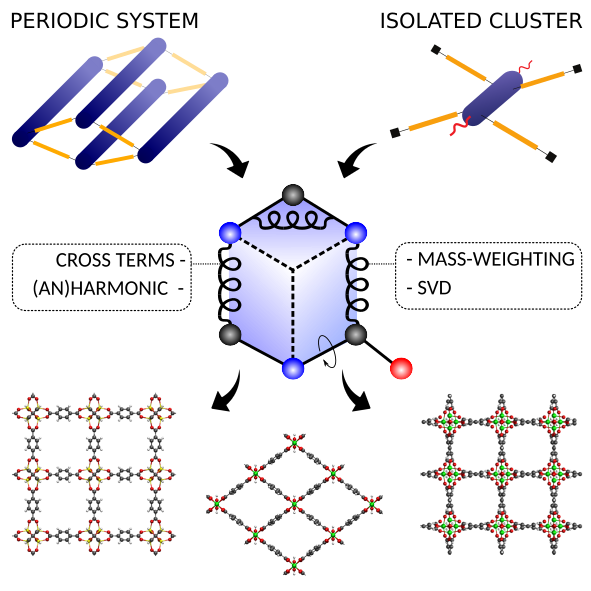
QuickFF was originally launched in 2015 to derive accurate force fields for isolated and complex molecular systems in a quick and easy way. Apart from the general applicability, the functionality was especially tested for metal-organic frameworks (MOFs), a class of hybrid materials consisting of organic and inorganic building blocks. Herein, we launch a new release of the QuickFF protocol which includes new major features to predict structural, vibrational, mechanical and thermal properties with greater accuracy, without compromising its robustness and transparant workflow. First, the ab initio data necessary for the fitting procedure may now also be derived from periodic models for the molecular system, as opposed to the earlier cluster-based models. This is essential for an accurate description of MOFs with one dimensional metal-oxide chains. Second, cross terms that couple internal coordinates (ICs) and anharmonic contributions for bond and bend terms are implemented. These features are essential for a proper description of vibrational and thermal properties. Third, the fitting scheme was modified to improve robustness and accuracy. The new features are tested on MIL-53(Al), MOF-5, CAU-13 and NOTT-300. As expected, periodic input data is proven to be essential for a correct description of structural, vibrational and thermodynamic properties of MIL-53(Al). Bulk moduli and thermal expansion coefficients of MOF-5 are very accurately reproduced by static and dynamic simulations using the newly derived force fields which include cross terms and anharmonic corrections. For the flexible materials CAU-13 and NOTT-300, the transition pressure is accurately predicted provided cross terms are taken into account.